https://www.jianshu.com/p/6a4241218fb5
先查看一下tbtools的付费插件
授权:https://www.jianshu.com/p/2a5f11f05146
介绍:https://mp.weixin.qq.com/s/AIUZforH1BIx4Lyru88kRQ
Advanced HMMer Search,从某个物种中鉴定某个家族的全部成员,输入PFAM ID,HMM文件,MSA,和蛋白质序列集合.pep文件,输出文件
插件 | 地表最强 Hmmer Search 界面工具,详细,有三种输入的实例
https://zhuanlan.zhihu.com/p/358660673
从某个物种中鉴定某个家族的全部成员
1,基于Domain,用hmmsearch等软件
2,基于序列相似性,用BLAST等软件

Newick Rename
常常有这种情况,用户会拿着一堆序列,构建一个进化树。花了时间,拿到了树文本,使用的时候才发现序列的 ID 不是自己想要的样式。于是最好的方法是…进行重命名。同样,这个需求来自半小时直播。我顺手就实现了,又是一众筹插件
https://mp.weixin.qq.com/s/yQCyVIheiJDxqnZ2R3ByEw

Batch MEME Motif SeqLogo Viz,一键可视化 MEME Motifs 组图
https://www.kpfans.com/article/3RWVekod9N.html


等,详细的在陈老师的网站里
Genome Region Comapre,
https://www.jianshu.com/p/5ce68f173174



BioPauGU
SRA to Fastq,转换格式
https://mp.weixin.qq.com/s/WC6Q1wr2M4CsdVZ2XYFjRA
FastOC,质量检测
https://mp.weixin.qq.com/s/Sz9enr_8s9P0goxEObn4TA
Trimmomatic,质控
https://mp.weixin.qq.com/s/Gmazcogi2KBNkv7J4hXh9Q
sKallisto Super Wrapper
https://mp.weixin.qq.com/s/zhYjsF-LiPzPetbVh7bfcA
Hisat2 Build Index
索引
Hisat2 Align
Stringtie Assembly, 转录组组装,合并
一共两个对应的是 Stringtie 两个主要功能:
Stringtie Assembly : 转录组组装
Stringtie Quantify : 转录本表达量估计
https://www.jianshu.com/p/1be4da428c81


对于组装一步,参数简单,用户只需要提供排序好的BAM文件即可,这些文件,事实上可以通过前几天推的 Hisat-build 和 Hisat-align 插件来获取。需要注意的有三点:
参考物种基因结构注释是可选的
设置输出目录而不是输出文件,因为组装时是单个bam文件单独组装,最后再进行一次merge,得到最终可用的 GTF 文件(具体Fasta序列提取,可直接使用 TBtools 的 GXF Sequence Extract,请参考公号前述推文)
并行线程数,应该注意,此处的并行线程数与Stringtie软件的线程数并不等价。(我调整了stringtie源码,编译并使其可以在windows下运行,但windows系统限制,所以只能单线程,且速度上不来。这没关系,我们可以多个文件同时组装,这样也就只需要使用stringtie的单线程模式,毕竟很多时候,我们并不可能做一个样品的转录组测序和组装 - PS:windows下可能会慢不少,比如6G的转录组,估计组装时间大概要到 1个小时,在我的笔记本上。不过同时跑2个,那么也是1个小时嘛..)
组装结果大体如下,作者做了两个实际数据的,

在liniux中
如果您有多个BAM文件并希望用StringTie进行转录组组装,同时希望合并它们以创建一个统一的转录组注释,可以按照以下步骤操作:
单独组装每个样本:
首先,对每个BAM文件分别运行StringTie来组装转录本。1
2
3stringtie -G <annotation.gtf> -o <output_sample1.gtf> <input_sample1.bam>
stringtie -G <annotation.gtf> -o <output_sample2.gtf> <input_sample2.bam>
...这将产生每个样本的GTF文件。
合并所有样本的转录组:
使用StringTie的--merge选项来合并所有单独样本的GTF文件。1
stringtie --merge -G <annotation.gtf> -o <merged_output.gtf> <list_of_gtf_files.txt>
其中
<list_of_gtf_files.txt>是一个文本文件,其中包含了之前步骤生成的所有GTF文件的路径,每行一个。例如,
list_of_gtf_files.txt内容如下:1
2
3output_sample1.gtf
output_sample2.gtf
...
这个合并步骤将基于所有提供的样本数据创建一个统一的转录组注释文件。
可选的估算转录本丰度:
使用合并后的GTF文件,您可以对每个样本重新运行StringTie来估算转录本的丰度。1
2
3stringtie -e -B -G <merged_output.gtf> -o <output_sample1.gtf> <input_sample1.bam>
stringtie -e -B -G <merged_output.gtf> -o <output_sample2.gtf> <input_sample2.bam>
...这里,
-e参数告诉StringTie仅估算合并GTF文件中存在的转录本的表达,-B参数用于生成用于后续基因表达水平分析的ballgown软件包的输入文件。
确保在实际使用命令之前替换尖括号中的占位符,使用您的文件和路径名。这些步骤将帮助您合并多个BAM文件的转录组组装结果,并可以用于进一步的表达量分析。
在StringTie中,-e 参数用于告诉程序仅估算已知的转录本的表达量。具体来说,当你使用 -e 参数时,StringTie将只使用在你提供的GTF文件(在这个例子中是<merged_output.gtf>)中注释过的转录本,而不会尝试去发现任何新的转录本。
这通常在你想要得到关于已知转录本的更精确的表达量估计时使用,特别是当你使用一个合并后的GTF文件,这个文件可能包含了从多个样本中得到的所有可能的转录本。使用 -e 参数可以防止StringTie在后续的分析中引入任何不确定性,因为它将不再搜索可能不存在或未被注释的转录本。
在表达量分析中,这个选项常用于准备数据以供后续分析,如差异表达分析等。
Stringtie Quantify·转录本表达量估计
https://www.jianshu.com/p/1be4da428c81

使用简单,用户需要的注意的几乎只有三点:
排序好的BAM文件
一个必须的基因结构注释文件,可以是gff3也可以是gtf,比如使用 StringTie Assembly插件整理的组装结果。
ReadLength, 这一参数即测序读长。因为StringTie计算的其实是Coverage,并没有读段计数这一步。常见的操作是简单地基于Coverage反推raw counts。于是read length成为必要

注意到,我前面做StringTie Assembly的时候设置了参考注释,所以AcoXXXX等ID为原始注释ID(事实上是没有被组装出来的,一般是覆盖率极低),而MSTRGXXXX等为已有注释中被当前样品覆盖以及新注释出来的基因。
PS:四个插件将会一并上线至插件商店,暂时票价应该会定位 100,打包估计 365,具体再定。
大概做法:每天看两三个TBTOOLS教程。(仅自学使用)
然后实操教程:
大概输入,出力,输出,有例子!
有陈老师的教程:https://www.yuque.com/cjchen/hirv8i/pso3pd
TBtools学会对分析很重要,所以专门来学习一下,并尝试用shinyapp代码实现相同的功能。

1,Fasta Extract (Recommended),知道ID和fa文件,提取出fa
https://www.yuque.com/cjchen/hirv8i/ra35nv
%E4%BD%8D%E7%BD%AE.png "TFasta Extract(Recommend)位置")

功能演示")
提取fa
另外:也能按照
#注意,制表符[\t]分隔,而非空白[Space]分隔
Chr1 10000 20000
#提取反向互补序列,则翻转碱基坐标
Chr1 20000 10000
1.1功能演示")
和下面这两种提取,输出后改名,手动改也行。好的我知道了
peak_1 Chr1 10000 10200
promoter_ATG8 Chr2 20300 22300
我平时知道ID和fa用linux为(很麻烦):
gffread -w transcripts.fa -g Gallus_gallus.GRCg6a.dna.toplevel.fa merge_2.gtf
gtf咋得?transcript_ID是id文件
grep -Ff transcript_ID -w mergelist原来.gtf > lncRNA.gtf
gffread protein_coding_gene.gtf -g /home/Gallus_gallus.GRCg6a.dna.toplevel.fa -w protein_coding_gene.fa
确实很方便,最后这个也很强大,OK不想复现。

2,Fasta Extract or Filter (Quick),知道ID和fa,获得fa
适合最多或极少
主要优势
相较于其他 Fasta 序列操作功能,这一功能的主要特点即,Quick。主要原因有二:
- 通过IO优化,加速文件读取
- 无需建立索引
适用场景
这一功能最适用的场景为 序列完整纪录 的 提取 或 过滤。
如果要问如何选择Fasta Extract (Recommended)与Fasta Extract or Filter (Quick)两个功能,可以看以下例子:若A基因组有两万个基因,想提取其中一万个基因,推荐使用Fasta Extract or Filter (Quick),若提取少量基因推荐用Fasta Extract (Recommended),若提取极少量基因推荐使用Fasta Extract or Filter (Quick)
Fasta Extract (Basic)
陈老师的情怀,用上面的提取就行
Fasta Stat 序列信息,获得fa文件的信息
https://www.yuque.com/cjchen/hirv8i/gm4yda
适用场景
很多时候,我们拿到一个 Fasta 序列文件,可能希望知道:
- 有多少个序列?
- 序列长度如何?序列总长度?
- 如果是DNA序列,GC含量如何?
- 每条序列信息指标,如长度,碱基组成等
- ….
Fasta Stat 功能可以快速完成上述统计信息。

选项解释如下:
- 输入序列文件(File Input)也可以直接黏贴序列(Seq Input)
- 待输出的序列整体概况
- 可选设置,可不设置;如设置具体输出文件,则会将每条序列的统计信息输出到该文件
- Keep Only Sequence Length: 若勾选,3. 中输出文件仅会包含序列ID及其长度(注意,这一功能主要是方便快速获取每条序列长度)
- 点击 Start 即可运行
Sequence Manipulate (Rev&Comp) ,获得序列反向,互补,大小
序列互补等 我也生成过
在日常分析过程中,我们常常需要进行序列操作,如序列反向,互补,大小写等。此功能为满足这些需求开发
确实,设计引物的时候

- 输入序列(以下三种方式)
● 拖入文件
● 粘贴部分序列
● 点击文本框后跟随的 “···”,在弹窗选择文件 - 输出文本区域框(设置好参数后自动输出,无需点击Start)
- 主要转换参数功能如下,勾选立即生效:
● Reverse:序列反向
● Complement:序列互补
● RNA:DNA序列转换成RNA序列
● Only IDs:仅保留ID(即去除序列)
● Only Seqs:仅保留序列(即去除所有ID)
● UpperCase:序列转换为大写
● LowerCase:序列转换为小写
● Seq in One Line:序列全部放在一行
● Base in Per Line:序列格式化为多行时,每行 60 个(与 Seq in One Line 冲突,若勾选Seq in One Line,此功能不起作用) - Clean:点击即清理输出
- Refresh:点击即可更新输出(一般用不上,当然如果你重新输入序列后,由于输出文本框中仍是上次序列处理后的输出结果,此时可直接点击Refresh)
感觉少了序列转换成蛋白质之类的,但是涌MERGE就行。
Fasta ID Simplify,fa文件id简化
有时候,一些 Fasta 序列 ID 较为复杂,如“>Unigene1 MYB Protein”,或从 NCBI,Uniprot,Swissprot 等公共数据库下载序列时,往往会遇到复杂 ID。这一功能主要用于简化 Fasta 序列 ID,例如:
“>Unigene1 MYB Protein”简化为“>Unigene1”。这功能也能专门针对一些数据库下载的特殊的 ID 格式。

使用详细解释(注意,该功能是批量的):
- 设置输入序列文件或直接黏贴少量序列文本
- 设置输出序列文件或直接输出到文本区域框
- Remove from:
从给定匹配模式处开始清理 ID,
如“>Unigene_1.version3.3 confident”,使用模式“.version”即可将清理为“>Unigene_1”;如“>Unigene1.transcript_01.version3.3 confident”,使用模式“3.3”即可将清理为“>Unigene1.transcript_01.version”
4. Remove .version:用于简单快速去除版本号等信息,如“>ABC183710.1”和“>CABT19912.3”,会直接去除“.1”和“.3”
5. 点击“Simplify My Sequence’s ID…”
这个应该也能写个shinyapp
Fasta ID Rename,fa文件重命名

参数详解如下:
- 设置输入的 Fasta 序列文件
- 设置输出文件路径
- 设置 ID 重命名映射信息,整体输入格式为(原始ID[\t]新ID),中间使用Tab键。注意,使用制表符分隔,。
- 点击Start即可运行
确实挺不错的
Fasta ID Prefix,给fa文件id增加前缀,方便统一
适用背景
在一些生信数据分析中,需要解决不同Fasta序列ID冲突的问题。如香蕉基因组序列染色体ID为“Chr1”“Chr2”…,而菠萝基因组序列染色体ID也为“Chr1”“Chr2”…。当我们在进行分析时,会存在一些软件无法正确处理的情况。最好的解决方法,就是对ID进行一定的调整,比如加前缀。

Fasta to Table Convert,fa格式不在同一格子,互换
Fasta 格式将 ID 和序列放置在不同行,在一些数据分析时,此功能可以将Fasta格式转换成 ID 和序列在同一行的表格模式,也可以将文件从 表格模式 转换成 Fasta 格式。


Merge and Split,对fa文件合并,或者分割
适用场景
在序列操作过程中,可能需要进行Fasta序列文件合并或者分割。大体场景可见:
- 多个 Fasta 序列文件合并,如Sanger测序得到的系列 .seq 文件
- 对含有多个序列的 Fasta 文件进行分割,如 10000 个序列,按照文件大小/序列数目/最终文件个数进行序列均分



序列文件合并
- 拖拽放置或者点击摁钮选择,设置多个待合并的序列文件
- 设置输出文件路径
- 点击 Start 即可开始文件合并
多序列文件分割
4. 设置输入的待分割的序列文件
5. 设置输出文件路径(注意是前缀路径,因为绝大多数情况下不止一个输出文件)
6. 设置分割单位数,结合 7. 考量
7. 设置分割单位,可选项为:
● Record Per File:即指定分割后每个文件的序列个数
● Number of File:即指定分割出来的结果文件数
● Size[KB] Per File:即指定分割出来的结果文件大小
8. 点击Start
Fasta Get Representative,多条序列的Fasta文件中提取出最长的代表性序列
适用场景
基于输入的 Fasta 序列文件 ID,提取代表性序列集合。即在一个有多条序列的Fasta文件中提取出最长的代表性序列,用于下游分析。
这个功能目前只建议在无法获得物种的 GFF3、GTF 文件和基因组序列信息的情况下使用。


Sequence Pattern Locate,掘简单重复序列
挖掘简单重复序列的
https://www.yuque.com/cjchen/hirv8i/zrdn6m


从mRNA中预测ORF(开放阅读框),ATG(?:\w{3})+T(?:AG|AA|GA),随后对每个序列取最长的一个ORF即为正确ORF
预测开放阅读框:
linux有个ORFfinder也能找到
ATCG 对应的就是四个碱基,ATCG
[ATCG] 对应的是一个碱基,A或T或C或G
[^AT] 对应的是一个碱基,但不会是A或T(将其排除)
(AT) 对应的是两个碱基,把AT定位为一个单元
(AT){6} 对应的是2x6,共12个碱基,也就是AT重复6次,ATATATATATAT
(AT){,6} 对应的是(AT)重复不多于6次,如AT,ATAT,ATATAT,ATATATAT,ATATATATAT,ATATATATATAT
(AT){6,} 对应的是(AT)重复不低于6次,如ATATATATATAT,ATATATATATATAT, ATATATATATATATAT等
SSR miner,快速挖掘并鉴定可用的 SSR 位点
基于输入的 Fasta 序列文件,快速挖掘并鉴定可用的 SSR 位点。

结果包含6列,内容分别是:
1.序列ID
2.起始坐标
3.终止坐标
4.SSR序列
5.重复单元长度(kb)
6.重复单元个数

Fasta Window Stat,对 fa序列进行滑窗统计,比如对基因组序列滑窗统计 GC偏斜,GC含量分布,N含量等
适用场景
对 Fasta 序列进行滑窗统计,比如对基因组序列滑窗统计 GC偏斜,GC含量分布,N含量等,看看基因组序列中未知碱基的分布(一般用 N 指代),以此来判断基因组不同区域的组装质量(是否有 Gap 等)。



NCBI Sequence fetch
GenBank to Fasta,GenBank格式转换成fa格式
从NCBI 下载序列时,可以保存为 GenBank 格式,从而保留尽可能多的序列信息。而有时候我们需要的是Fasta格式的序列,使用这一功能可以将GenBank格式的序列转化为Fasta格式。



NCBI Sequence Download (Basic),知道NCBI的Accession ID或者GI号,下载序列
适用场景
下载NCBI序列。但这一功能不建议一次下载超过100个序列,如有该需求,可以使用Bulk NCBI Sequence Download (Advanced)功能。



Bulk NCBI Sequence Download (Advanced),批量下载NCBI序列

ORF Predict
ord
Complete/Partial ORF Predict (Single Mode),从一段转录本(mRNA)序列上预测六个读码框的可能完整 CDS
https://www.yuque.com/cjchen/hirv8i/cktn7p
可以从弹出的窗口中看到:
- 按照 6 个读码框翻译的所有可能 ORF
- 点击灰色部分,即可查看该 ORF 信息(CDS 和 蛋白序列)
- 详细的ORF信息
● 预测的 ORF 信息(起始坐标,终止坐标,框架信息,长度等)
● >nt:CDS 序列
● >aa:氨基酸序列
支持不完整 ORF
版本更新后,支持非完整 ORF,比如缺少翻译起始密码子或者缺少终止密码子,更或者两者都缺少。当然,同时是支持非经典起始或终止密码子。
仍然是同样的数据,不同的参数
- 输入文本框
- 其他参数
● startCodon:若不勾选,则预测出来的ORF结果不包括起始密码子
● stopCodon:若不勾选,则预测出来的ORF结果不包括终止密码子
● Min Pep Len:最小氨基酸序列长度 - 点击Get ORF
Complete ORF Prediction (Batch),批量从一段转录本(mRNA)序列上预测六个读码框的可能完整 CDS
https://www.yuque.com/cjchen/hirv8i/gc37kd
Single 模式的 ORF 预测功能仅支持一条序列的预测,而有时候我们需要对一个序列基因进行批量预测,如5w个基因的转录本信息。此时可使用Complete ORF Prediction (Batch)这一功能
Batch Translate CDS to Protein,这一功能的作用是,按照标准密码子表,批量转换 CDS 为 蛋白序列
https://www.yuque.com/cjchen/hirv8i/hdr91x
这一功能的作用是,按照标准密码子表,批量转换 CDS 为 蛋白序列
Six Frame Translate,存在一些情况,我们希望对输入序列,进行六框翻译。
https://www.yuque.com/cjchen/hirv8i/ecsea1zqebf69vns
Primer Check (Simple e-PCR),检查设计的引物是否真能在fa上
https://chat.openai.com/g/g-l38NcMokB-mia-ai-your-voice-ai-companion/c/dc9aeb77-04ad-4c71-b023-2587ea7e82b6
适用场景
当前,几乎任何一个物种都可以简单获得转录本序列集合,于是我们在设计 q-PCR 荧光定量引物后,完全可以比对到这一序列库,查看是否会产生非特异性扩增,可用于简单的PCR引物特异性检测。
ps:本功能是早期开发TBtools时写的,用的是最简单的字符串搜索和编辑距离计算方法,有运行速度慢且未考虑热力学结果,仅仅基于字符串匹配与否等缺点
建议使用改进后的“Prime Check”插件
操作流程
引物设计:根据目标基因序列设计一对引物。
序列获取:收集目标物种的全部转录本序列,这些序列可以从公共数据库如NCBI、Ensembl等获取。
比对分析:使用Simple e-PCR或类似的生物信息学工具,将设计的引物与转录本序列库进行比对分析。
结果分析:
特异性匹配:理想情况下,引物仅与目标序列产生特异性匹配,不与其他非目标序列匹配,这意味着引物设计是成功的。
非特异性匹配:如果引物还与其他非目标序列产生匹配,这可能导致非特异性扩增,此时需要重新设计引物。
结果应用
优化引物设计:基于比对结果,重新设计引物,避免潜在的非特异性扩增。
提高实验准确性:确保q-PCR实验的准确性和可靠性,通过特异性的引物扩增来获得准确的定量结果。
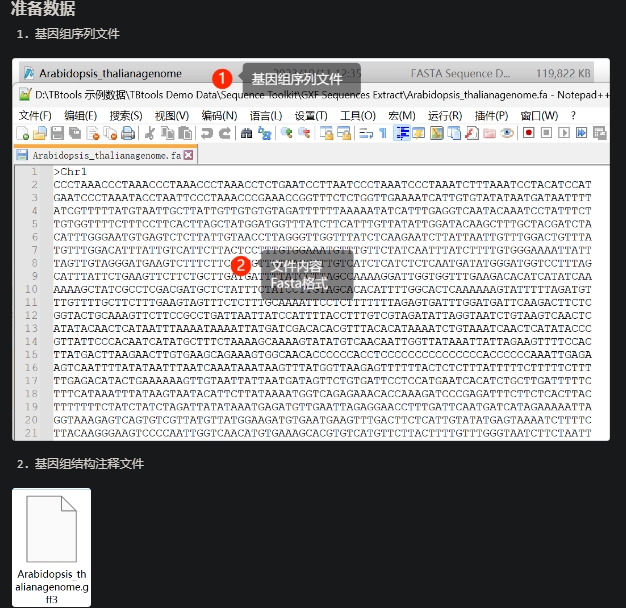
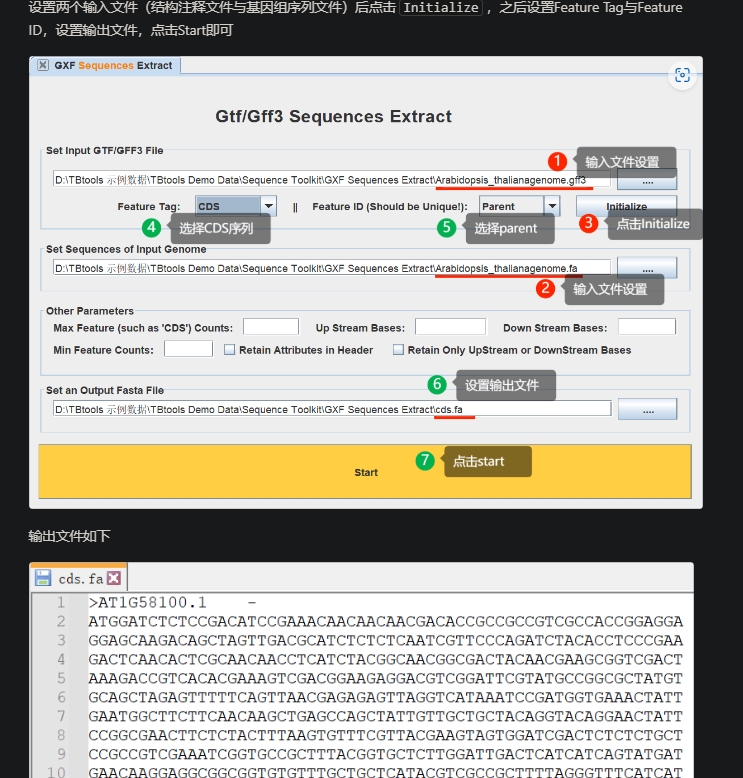
GXF Sequence Extract,输入fa,gff,得到fa文件
https://www.yuque.com/cjchen/hirv8i/olq9pq
适用场景
目前,不少人物种的基因组已经被测定,常常我们可以直接获得物种的基因组序列以及基因结构注释信息。基于这两个文件,我们完全可以提取:
● 转录本序列
● CDS序列
● 启动子序列
● …
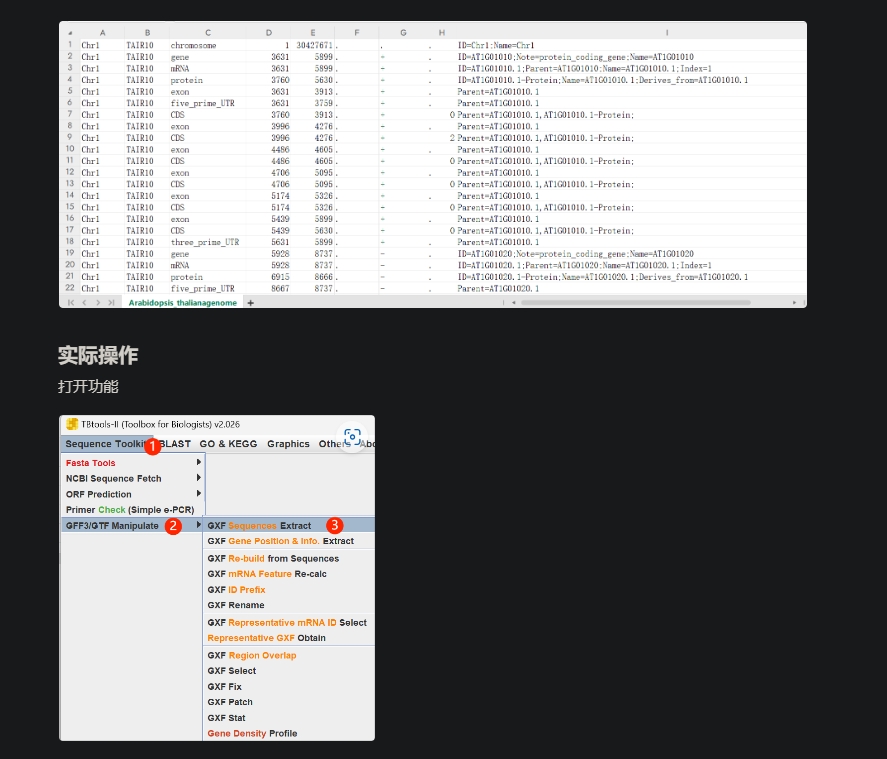

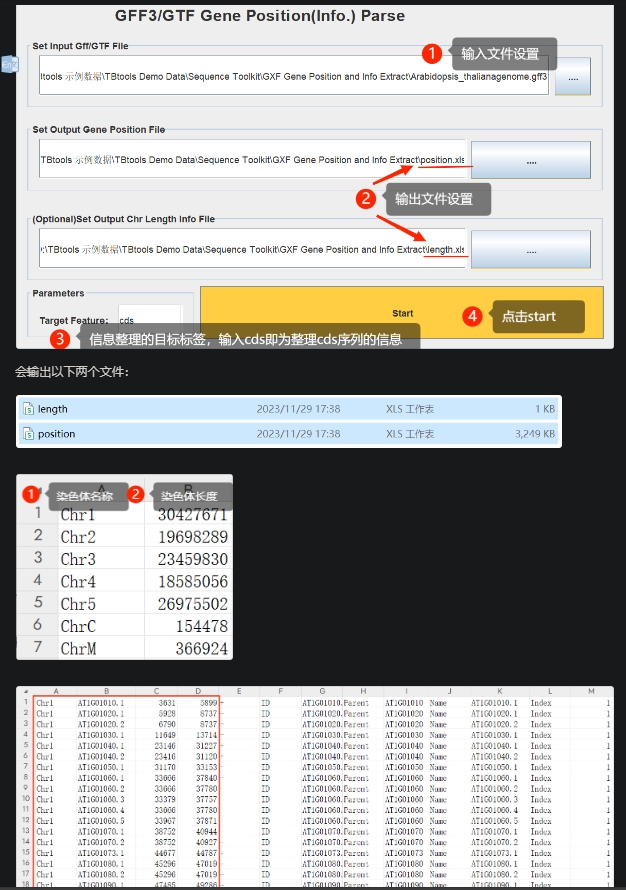
GXF Gene Position & Info Extract,中提取每个基因的具体信息,如位置,注释等
https://www.yuque.com/cjchen/hirv8i/gbsdn6
物种基因结构注释信息文件,GFF3/GTF记录了全面的物种功能元件信息。有些时候,我们希望从中提取每个基因的具体信息,如位置,注释等。
这一功能可以帮助我们快速获取这些信息
GXF Re-build from Sequence,直接基于转录本序列或CDS和基因组序列文件,直接重构 GFF3 文件。
https://www.yuque.com/cjchen/hirv8i/ota5q6
适用场景
尽管很多物种都公开了物种基因结构注释信息,但仍然存在不少有问题的基因结构注释,更或者有一些基因结构没注释出来,需要补充。那么简单的办法,即直接基于转录本序列或CDS和基因组序列文件,直接重构 GFF3 文件。
TBtools | 基因组重测序数据分析常用上游软件「三兄弟」,聚齐了~
https://www.jianshu.com/p/a98b3b69bcfe
软件教程 | 利用TBtools完成无门槛构建系统发育树的全套流程
完结 | TBtools RNAseq 数据分析系列插件
https://www.jianshu.com/p/c8b08314e133
整体上,覆盖了数个功能,四个插件:
1,SRA 数据查询与整理:SRA XML to Table,见推文:挖掘SRA的辅助小工具(NCBI高通量测序数据收录库)https://mp.weixin.qq.com/s/FnuSUqhpyKqm_HYpu6phnw
2,SRA 数据链接获取:SRA XML to Table 和 SRA Number to ENA Info. 前者已经包括了 NCBI 和 DDBJ 数据下载链接,后者主要作为补充,附加 ENA 下载链接(更为稳点)。详细见:公开可获取~没有下载不到的测序原始数据!https://mp.weixin.qq.com/s/CS04e0QRjq0B-NZUfCpUAg
3,Ascp GUI Wrapper:个人实测,每天清晨通过 FTP 链接下载测序原始数据,速度可以达到 10Mb/s。但更多时候数据只有不到 300Kb/s。网络合适的情况下,可以使用 Aspera ,速度可以达到 30Mb/s。于是写了并公开释放了这个插件,详细见:插件 | 人人-点点点-光速下载 NCBI/ENA NGS原始数据 https://mp.weixin.qq.com/s/YYneVPb3V6Dq5WXiq2JYTQ
4,SRAtoFastq,sra 是 NCBI 定义的二代数据存储格式,文件大小比fastq.gz下,考虑网络带宽的情况下,下载 sra 数据更方便。下载后需要进行转换,于是有了插件,详细见:SRAtoFastq | 任何人都能自主分析测序原始数据 https://mp.weixin.qq.com/s/WC6Q1wr2M4CsdVZ2XYFjRA
5,FastQC,无论是NCBI SRA等数据库下载,还是公司返还的测序数据,多少还是要看下测序质量,确保质量OK 或者不要有样品降解,严重污染云云,于是有插件,详细见:插件FastQC | 点点点,人人看看测序数据质量 https://mp.weixin.qq.com/s/Sz9enr_8s9P0goxEObn4TA
6,Trimmomatic,无论转换得到,或者是公司测序后返还的 Fastq.gz 数据往往是原始数据,通过 FastQC 可以判断,随后进行质量控制,如去除接头和低质量碱基,于是有插件,详细见:Trimmomatic | 点点点,测序原始数据质控,技能√get https://mp.weixin.qq.com/s/Gmazcogi2KBNkv7J4hXh9Q
7,Kallisto,RNAseq 数据的基本分析和目的,就是获得基因表达量矩阵。在普通笔记本上,如 4G 内存云云,那么 Kallisto 是最好的选择,于是有插件,详细见:
Kallisto | 点点点,从 测序数据 到 基因表达量矩阵 人人都可以! https://mp.weixin.qq.com/s/zhYjsF-LiPzPetbVh7bfcA
Trans Value Sum,Kallisto 分析结果是转录本水平的表达量或Counts矩阵,但很多人感兴趣的是基因水平的,于是,公开释放了功能,详细见:汇总 | 转录本表达矩阵 到 基因表达矩阵 https://mp.weixin.qq.com/s/JPM7ofuqZcKPZjySL7w5lA
至此,完结。
因为接下来的步骤,那么无非是:
差异表达分析,拿到Counts 矩阵后,可以用我齐哥的网页工具(后续写个教程贴)
富集分析(GUI 的,基于最常用的,直接使用 TBtools 使用手册,几年前已经个释放了)
… 暂时没想出其他通用的分析步骤…
作者:生信石头
链接:https://www.jianshu.com/p/c8b08314e133
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。



